
Identifying patients who are eligible for clinical trials is one of the fundamental challenges the cancer research community faces. While there are several personal reasons that may dissuade a patient from participating in research, there are also many logistical barriers—identifying a patient at just the right time, when they are ready to be put on a therapy but have not yet started one, is often challenging when a practice may have dozens of trials open, each with a dozen or more inclusion/exclusion criteria, and with hundreds of patients coming into a practice a day.
This is where we think technology can help. However, people with software experience from other industries often assume the answer is one of two extremes: either a complex ensemble of ML approaches or a simple SQL statement comparing the patient data to the trial's requirements. Unfortunately, the data available is typically dirty, incomplete, unlabeled, and complex. There is inherently no 100% right answer, no matter how sophisticated your approach because you're never given 100% of the data needed to solve the problem. The "right" answer is any that performs better than the current solution, provides visibility into its workings, and could even be one you learned in CS 101.
When looking at a clinical trial, there are many different potential eligibility criteria: diagnosis, stage, age, prior therapies, and biomarkers are among the most common. As we've matched patients in the past, we've thought about this as a linear pathway:
-
Does the patient match on disease? If yes, go to step 2. If not, they are not eligible.
-
Does the patient have the correct biomarker mutations? If yes, go to step 3. If not, they are not eligible.
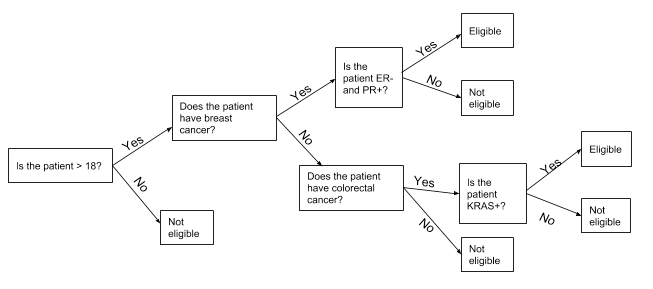
As we started to look at adding more criteria, and specifically biomarkers—measurable, biological indicators related to disease—we started to run into an issue: most trials were not so straightforward, and our current process for adding criteria wasn't going to hold up. Take the very simplified criteria for a real multi-tumor study (across several diseases) with a few different biomarkers (in this case, ER, PR and KRAS):
-
Is the patient over 18? If yes, go to step 2. If not, they are not eligible.
-
Does the patient have breast cancer? If yes, go to step 3. If not, go to step 4.
-
Is the patient ER- and PR-? If yes, they are eligible. If not, they are not.
-
Does the patient have colorectal cancer? If yes, go to step 5. If not, they are not eligible.
-
Does the patient have a KRAS mutation? If yes, they are eligible. If not, they are not.
As we started to run into this problem, we participated in the TOP Health sprint to improve our trial-to-patient matching capabilities. Through this, we had access to a mock dataset which had been hand-curated by the National Cancer Institute. In this dataset, they structured their trial criteria as boolean expression.
For the one above, it might be represented as:
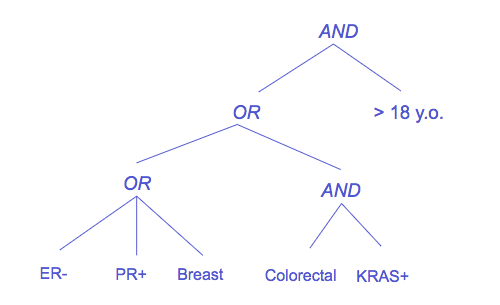
(Age > 18) AND (ER = Negative AND PR = Negative AND Disease = Breast) OR (KRAS = Positive AND Disease = Colorectal)
This led to a breakthrough—instead of storing each eligibility criteria type in its own silo ("Here are the disease criteria, here are the biomarker criteria"), what if they could be stored and evaluated in a common data structure? If we treat the boolean operators above as if they are mathematical expressions, we can create a classic expression tree (internally, we've been calling this a decision tree, but that's something else).
Taking the example above, we can translate that into the tree on the right below:
When we evaluate each node, we bubble up its result to the node above it, eventually getting to a result of whether a patient is eligible for this trial.
In the example below, the patient has breast cancer and is ER- and PR-, but doesn't have colorectal cancer and hasn't tested KRAS+. Despite that, they are eligible for the trial because of the left subtree.
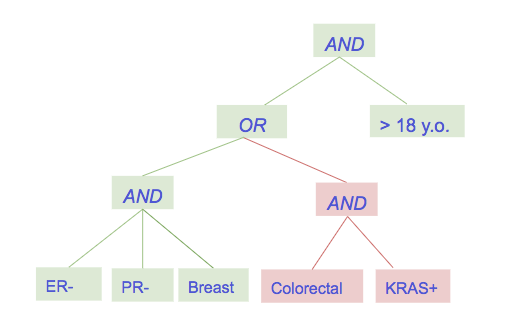
Each leaf node in the Flatiron clinical trials decision tree represents a single inclusion or exclusion criterion. The nodes (and their criteria) are mixed and matched into different trees to form the criteria for different trials. In the TOP Health sprint, we parsed experimental criteria expressions to form these trees, and supplemented it with data from clinicaltrialsapi.cancer.gov. As we move this into production, the eligibility criteria isn't clean enough for us to confidently use it in these mappings, so we are going to work with our partner sites to input the criteria for the trials open at their sites.
However, we've found that one of the biggest impediments to effective trial matching is the lack of trial criteria information, meaning that we've been limited in their ability to suggest trials for which a patient is actually eligible. With the comprehensive trial information provided for the sample dataset in TOP Health, we were able to greatly improve our ability to match attributes on the mock patients. If the cancer.gov trials API included something similar to eligibility expressions for each trial, it could create a template for both patient-to-trial and trial-to-patient matching that developers across the industry could follow, and would have an enormous benefit for patients.
Using an expression tree preserves the visibility and explainability that comes with a deterministic matching approach while allowing us to query entirely disparate data sources, with match rankings, through a unified interface. Moving forward, this also enables us to incorporate machine learning into our approach, having nodes return probabilities instead of absolutes.
When written out, the approach is nothing complex. Each leaf node has a job—for instance, it knows how to take a patient's clinical information and return whether the patient matches on disease. Adding a new type of criteria involves only adding a new type of leaf node. When you consider that each node has the freedom to do something as complex as call out to a machine learning model for help making its decision, it's tempting to use words like "system" or "ensemble coordinator" but… it's still just a tree. A simple data structure that can help solve complex problems with the right nurturing.
Appendix: Runnable Code
In an effort to make this post more concrete, we've included a simplified version of our matching tree code below to show how it works together. There are two "Operator" classes (AND and OR), which can have children added to them, and two "Leaf" classes.
For the sake of simplicity, our equivalent of a patient is MockMatchable, a namedtuple that has a number and a series of letters associated with it. One leaf class, LetterMatchLeaf, only allows a certain subset of letters. The other, NumberMatchLeaf, only allows numbers less than a certain number. In our production system, we have leaves such as DiseaseMatchLeaf and BiomarkerMatchLeaf, but the effect is the same.
At the bottom of the code, we build a simple tree and evaluate different MockMatchable objects against it. The tree could return probabilities, but in our simple example, it only returns 1 or 0. Our nonsensical tree looks like this:
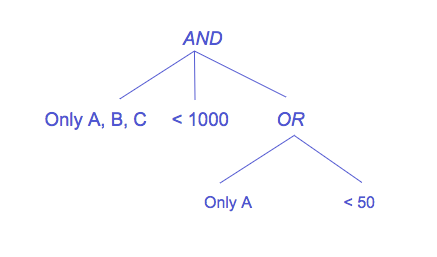
We'd encourage you to play around building different trees, new leafs, and MockMatchable objects to see how we can build upon this.
Disclaimer: There are many ways to improve this code, but in the interest of brevity, we've kept it short—please ignore the lack of real inheritance and other things you might expect in production code.


